RNA correlated boundary#
import numpy as np
import pandas as pd
import cooler
import anndata
import scanpy as sc
import matplotlib as mpl
import matplotlib.pyplot as plt
from matplotlib.colors import LogNorm
from matplotlib import cm as cm
import seaborn as sns
mpl.style.use('default')
mpl.rcParams['pdf.fonttype'] = 42
mpl.rcParams['ps.fonttype'] = 42
mpl.rcParams['font.family'] = 'sans-serif'
mpl.rcParams['font.sans-serif'] = 'Helvetica'
indir = '/data/hba/domain_majortype/'
outdir = '/home/jzhou_salk_edu/sky_workdir/hba/rna_majortype/'
# no L5ET
leg = ['L23_IT', 'L4_IT', 'L5_IT', 'L6_IT', 'L6_IT_Car3', 'L56_NP', 'L6_CT', 'L6b', 'Amy',
'Lamp5', 'Lamp5_LHX6', 'Sncg', 'Vip', 'Pvalb', 'Pvalb_ChC', 'Sst', 'CHD7',
'MSN_D1', 'MSN_D2', 'Foxp2', 'SubCtx',
# 'ASC', 'ODC', 'OPC', 'MGC', 'PC', 'EC', 'VLMC'
]
res = 25000
bound_count_ct = pd.read_hdf(f'{indir}MajorType_boundcount.hdf')
cell_count_ct = pd.read_csv(f'{indir}MajorType_cellcount.csv.gz', header=0, index_col=0)['count_cell']
bound_prob_ct = bound_count_ct.loc[leg] / cell_count_ct.loc[leg][:, None]
expr = pd.read_hdf('/home/jzhou_salk_edu/sky_workdir/hba/rna_majortype/cluster_expr.hdf').loc[leg]
from scipy.stats import rankdata
deg = np.zeros(expr.shape[1])
for i in range(len(leg)-1):
for j in range(i+1, len(leg)):
tmp = np.load(f'/home/jzhou_salk_edu/sky_workdir/hba/rna_majortype/DEG/{leg[i]}-{leg[j]}.npz')
# deg[np.logical_and(np.abs(tmp['fc'])>1, tmp['fdr']<1e-3)] = 1
rank = rankdata(tmp['fdr'])
deg[rank<=100] = 1
print(deg.sum())
1131.0
chrom_size_path = f'/data/hba/loop_majortype/hg38_with_chrl.main.chrom.sizes'
chrom_sizes = cooler.read_chromsizes(chrom_size_path, all_names=True)
gene_meta = pd.read_csv('/home/jzhou_salk_edu/sky_workdir/hba/ref/gencode.v33.bed', sep='\t', index_col=None, header=None)
gene_meta.columns = ['chrom', 'start', 'end', 'gene_id', 'gene_name', 'strand']
gene_meta = gene_meta.set_index('gene_id').loc[expr.columns[deg==1]]
gene_meta = gene_meta.loc[gene_meta['chrom'].isin(chrom_sizes.index[:22])]
gene_meta
| chrom | start | end | gene_name | strand | |
|---|---|---|---|---|---|
| gene | |||||
| ENSG00000002746.15 | chr7 | 43112629 | 43566001 | HECW1 | + |
| ENSG00000005108.16 | chr7 | 11370365 | 11832198 | THSD7A | - |
| ENSG00000006128.12 | chr7 | 97732084 | 97740472 | TAC1 | + |
| ENSG00000006468.14 | chr7 | 13891229 | 13991425 | ETV1 | - |
| ENSG00000007237.18 | chr17 | 9910609 | 10198551 | AC005747.1 | - |
| ... | ... | ... | ... | ... | ... |
| ENSG00000286954.1 | chr6 | 22663507 | 22675493 | AL033539.2 | + |
| ENSG00000287172.1 | chr2 | 76185020 | 76399490 | AC073091.3 | + |
| ENSG00000287694.1 | chr16 | 76277288 | 76819624 | AC106741.2 | + |
| ENSG00000287722.1 | chr13 | 53207831 | 53801489 | AL356295.1 | + |
| ENSG00000287912.1 | chr11 | 81175201 | 81431520 | AP003464.1 | + |
1099 rows × 5 columns
binall = pd.DataFrame(index=bound_count_ct.columns)
binall['chrom'] = binall.index.str.split('_').str[0]
binall['start'] = binall.index.str.split('_').str[1].astype(int) * res
binall['end'] = binall['start'] + res
binall
| chrom | start | end | |
|---|---|---|---|
| chr1_0 | chr1 | 0 | 25000 |
| chr1_1 | chr1 | 25000 | 50000 |
| chr1_2 | chr1 | 50000 | 75000 |
| chr1_3 | chr1 | 75000 | 100000 |
| chr1_4 | chr1 | 100000 | 125000 |
| ... | ... | ... | ... |
| chr22_2028 | chr22 | 50700000 | 50725000 |
| chr22_2029 | chr22 | 50725000 | 50750000 |
| chr22_2030 | chr22 | 50750000 | 50775000 |
| chr22_2031 | chr22 | 50775000 | 50800000 |
| chr22_2032 | chr22 | 50800000 | 50825000 |
115009 rows × 3 columns
import joblib
# from qnorm import quantile_normalize
from scipy.stats import norm
from statsmodels.sandbox.stats.multicomp import multipletests as FDR
from tqdm import tqdm
from ALLCools.mcds.correlation import corr_array
bkl = pd.read_csv(f'/data/hba/loop_majortype/M1C.rowsumpb1000.blf50.merged.bed', sep='\t', header=None, index_col=None)
binall['bklfilter'] = True
for c in chrom_sizes.index[:-1]:
chrfilter = (binall['chrom']==c)
tmp = binall.loc[chrfilter]
tmp.iloc[:10, -1] = False
tmp.iloc[-10:, -1] = False
for xx,yy in bkl.loc[bkl[0]==c, [1,2]].values // res:
tmp.iloc[max([0,xx-2]):(yy+2), -1] = False
binall.loc[chrfilter] = tmp.copy()
print(binall['bklfilter'].sum())
100643
bound_prob_ct = bound_prob_ct.loc[:, binall['bklfilter']]
binall = binall.loc[binall['bklfilter']]
def shuffle_corr_norm(rna_data, dmr_data):
shuffle_rna_data = rna_data.copy()
for col, data in shuffle_rna_data.items():
n_gene = shuffle_rna_data.shape[0]
shuffle_rna_data[col] = shuffle_rna_data[col].sample(n_gene).values
if dmr_data.shape[0] > 50000:
shuffle_dmr_data = dmr_data.sample(50000).copy()
else:
shuffle_dmr_data = dmr_data.copy()
for col, data in shuffle_dmr_data.items():
n_dmr = shuffle_dmr_data.shape[0]
shuffle_dmr_data[col] = shuffle_dmr_data[col].sample(n_dmr).values
# shuffle corr
shuffle_corr = corr_array(shuffle_rna_data, shuffle_dmr_data)
mu, std = norm.fit(shuffle_corr.ravel())
return mu, std, shuffle_corr.ravel()
null_mu, null_std, shuffle_corr = shuffle_corr_norm(expr.loc[:, expr.columns.isin(gene_meta.index)].T, bound_prob_ct.T)
null_mu, null_std
(0.0019941576523659988, 0.2235632728434467)
shuffle_corr.shape
(54950000,)
gene_slop = 5000000
gene_records = []
for gene, row in tqdm(gene_meta.iterrows(), total=gene_meta.shape[0]):
gene_rna = expr[[gene]].T
dmr_chrom = row['chrom']
dmr_start = row['start'] - gene_slop
dmr_end = row['end'] + gene_slop
sel_dmr = (binall['chrom']==dmr_chrom) & (binall['start'] > dmr_start) & (binall['start'] < dmr_end)
gene_dmr = bound_prob_ct.T.loc[sel_dmr]
gene_corr = corr_array(gene_rna, gene_dmr).ravel()
gene_corr = pd.Series(gene_corr, index=gene_dmr.index)
# pvalue = norm.sf(gene_corr.values, null_mu, null_std)
# pvalue[pvalue > 0.5] = 1 - pvalue[pvalue > 0.5]
# pvalue *= 2 # two tailed
# perform multi-test correction and q-value
# _, q, *_ = fdrcorrection(pvalue)
gene_corr.name = 'corr'
gene_corr = gene_corr.reset_index()
gene_corr['gene'] = gene
# gene_corr["q"] = q
# minimum filter
# gene_corr = gene_corr[
# (gene_corr["q"] < min_q) & (gene_corr["corr"].abs() > min_corr)
# ].set_index("dmr")
# gene_records[gene] = gene_corr
gene_records.append(gene_corr)
100%|██████████| 1099/1099 [00:21<00:00, 51.51it/s]
gene_records = pd.concat(gene_records, axis=0)
gene_records.index = np.arange(gene_records.shape[0])
gene_records
| index | corr | gene | |
|---|---|---|---|
| 0 | chr7_1525 | 0.044345 | ENSG00000002746.15 |
| 1 | chr7_1526 | -0.298720 | ENSG00000002746.15 |
| 2 | chr7_1527 | -0.408159 | ENSG00000002746.15 |
| 3 | chr7_1528 | -0.556742 | ENSG00000002746.15 |
| 4 | chr7_1529 | -0.246539 | ENSG00000002746.15 |
| ... | ... | ... | ... |
| 424261 | chr11_3453 | -0.259785 | ENSG00000287912.1 |
| 424262 | chr11_3454 | -0.141301 | ENSG00000287912.1 |
| 424263 | chr11_3455 | 0.278937 | ENSG00000287912.1 |
| 424264 | chr11_3456 | 0.347317 | ENSG00000287912.1 |
| 424265 | chr11_3457 | 0.187560 | ENSG00000287912.1 |
424266 rows × 3 columns
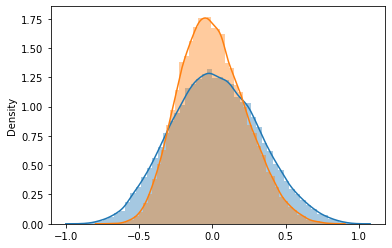
fig, ax = plt.subplots()
sns.distplot(np.random.choice(gene_records['corr'], 50000), ax=ax)
sns.distplot(np.random.choice(shuffle_corr, 50000), ax=ax)
<AxesSubplot:ylabel='Density'>
findfont: Font family ['sans-serif'] not found. Falling back to DejaVu Sans.
findfont: Generic family 'sans-serif' not found because none of the following families were found: Helvetica
t1 = rankdata(np.concatenate((gene_records['corr'].values, shuffle_corr)))[:gene_records.shape[0]]
t2 = rankdata(gene_records['corr'].values)
gene_records['FDRneg'] = (t1 - t2) / len(shuffle_corr) / t2 * gene_records.shape[0]
gene_records['FDRpos'] = (len(shuffle_corr) - t1 + t2) / len(shuffle_corr) / (gene_records.shape[0] - t2) * gene_records.shape[0]
threspos = gene_records.loc[gene_records['FDRpos']<0.1, 'corr'].min()
thresneg = gene_records.loc[gene_records['FDRneg']<0.1, 'corr'].max()
print(threspos, thresneg)
0.681722215979567 -0.514844193782862
gene_meta[['TSS', 'TES']] = gene_meta[['start', 'end']]
selg = (gene_meta['strand']=='-')
gene_meta.loc[selg, ['TSS', 'TES']] = gene_meta.loc[selg, ['TES', 'TSS']].values
gene_records['TSSdist'] = binall.loc[gene_records['index'], 'start'].values - gene_meta.loc[gene_records['gene'], 'TSS'].values
selg = (gene_meta.loc[gene_records['gene'], 'strand']=='-')
gene_records.loc[selg.values, 'TSSdist'] = -gene_records.loc[selg.values, 'TSSdist'].values
gene_records['TESdist'] = binall.loc[gene_records['index'], 'start'].values - gene_meta.loc[gene_records['gene'], 'TES'].values
selg = (gene_meta.loc[gene_records['gene'], 'strand']=='-')
gene_records.loc[selg.values, 'TESdist'] = -gene_records.loc[selg.values, 'TESdist'].values
gene_records['coord'] = 0
selp = (gene_records['TSSdist']<=0)
gene_records.loc[selp, 'coord'] = gene_records.loc[selp, 'TSSdist'] / res - 100
selp = (gene_records['TESdist']>=0)
gene_records.loc[selp, 'coord'] = gene_records.loc[selp, 'TESdist'] / res + 100
selp = (gene_records['TESdist']<0) & (gene_records['TSSdist']>0)
gene_records.loc[selp, 'coord'] = gene_records.loc[selp, 'TSSdist'] / (gene_records.loc[selp, 'TSSdist'] - gene_records.loc[selp, 'TESdist']) * 200 - 100
gene_records.to_hdf(f'{outdir}DEG_neu_bound_5M_corr.hdf', key='data')
gene_records = pd.read_hdf(f'{outdir}DEG_neu_bound_5M_corr.hdf', key='data')
thres = np.max(np.abs([thresneg, threspos]))
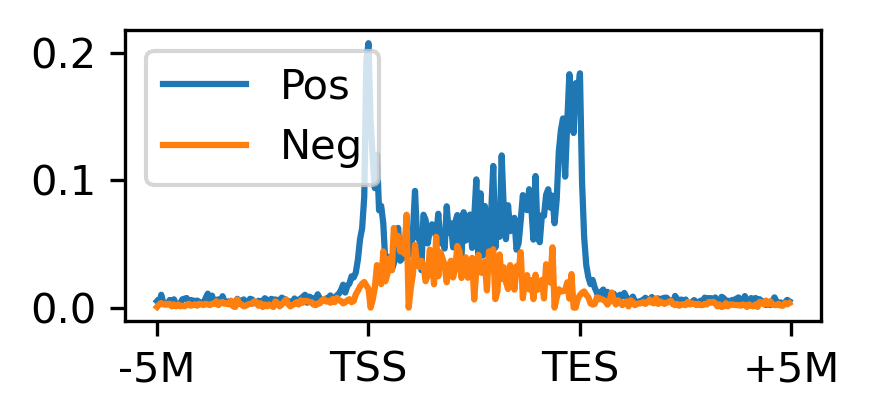
gene_records['group'] = gene_records['coord']//2
proppos = (gene_records.loc[gene_records['corr']>thres, 'group'].value_counts() / gene_records['group'].value_counts()).sort_index().fillna(0)
propneg = (gene_records.loc[gene_records['corr']<-thres, 'group'].value_counts() / gene_records['group'].value_counts()).sort_index().fillna(0)
fig, ax = plt.subplots(figsize=(3, 1.5), dpi=300)
ax.plot(np.arange(300), proppos, c='C0', label='Pos')
ax.plot(np.arange(300), propneg, c='C1', label='Neg')
ax.set_xticks([0, 100, 200, 300])
ax.set_xticklabels(['-5M', 'TSS', 'TES', '+5M'])
ax.legend()
plt.tight_layout()
# plt.savefig('proportion_corr_bound.pdf', transparent=True)
tmp = gene_records.loc[(gene_records['corr']>thres) & (gene_records['TSSdist']>-res) & (gene_records['TSSdist']<res), 'gene'].unique()
print(tmp.shape[0], tmp.shape[0]/gene_records['gene'].unique().shape[0])
np.savetxt(f'{outdir}gene_boundposcorr_tss.csv.gz', tmp, delimiter='\n', fmt='%s')
tmp = gene_records.loc[(gene_records['corr']>thres) & (gene_records['TESdist']>-res) & (gene_records['TESdist']<res), 'gene'].unique()
print(tmp.shape[0], tmp.shape[0]/gene_records['gene'].unique().shape[0])
np.savetxt(f'{outdir}gene_boundposcorr_tes.csv.gz', tmp, delimiter='\n', fmt='%s')
tmp = gene_records.loc[(gene_records['corr']>thres) & (((gene_records['TSSdist']>-res) & (gene_records['TSSdist']<res)) | ((gene_records['TESdist']>-res) & (gene_records['TESdist']<res))), 'gene'].unique()
print(tmp.shape[0], tmp.shape[0]/gene_records['gene'].unique().shape[0])
np.savetxt(f'{outdir}gene_boundposcorr_tsstes.csv.gz', tmp, delimiter='\n', fmt='%s')
285 0.25932666060054593
271 0.24658780709736125
466 0.4240218380345769
tmp = gene_records.loc[(gene_records['corr']>thres) & (gene_records['TSSdist']>-res) & (gene_records['TESdist']<res), 'gene'].unique()
print(tmp.shape[0], tmp.shape[0]/gene_records['gene'].unique().shape[0])
np.savetxt(f'{outdir}gene_boundposcorr_genebody.csv.gz', tmp, delimiter='\n', fmt='%s')
tmp = gene_records.loc[(gene_records['corr']<-thres) & (gene_records['TSSdist']>-res) & (gene_records['TESdist']<res), 'gene'].unique()
print(tmp.shape[0], tmp.shape[0]/gene_records['gene'].unique().shape[0])
tmp = gene_records.loc[(gene_records['corr'].abs()>thres) & (gene_records['TSSdist']>-res) & (gene_records['TESdist']<res), 'gene'].unique()
print(tmp.shape[0], tmp.shape[0]/gene_records['gene'].unique().shape[0])
591 0.5377616014558689
224 0.20382165605095542
674 0.6132848043676069
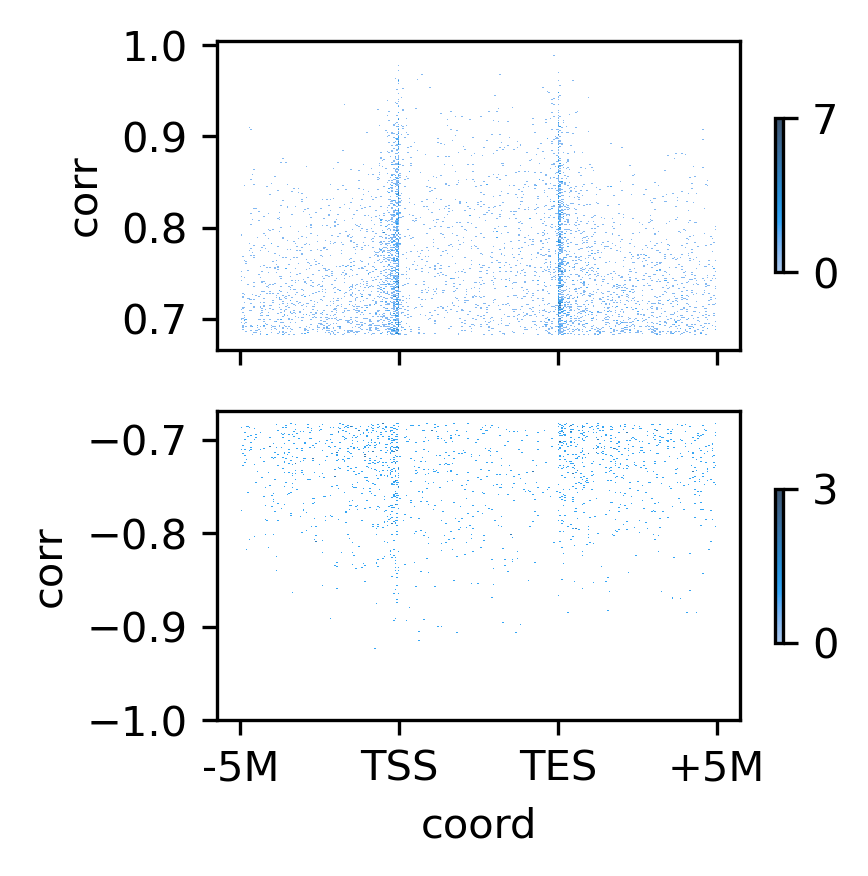
fig, axes = plt.subplots(2, 1, sharex='all', figsize=(3,3), dpi=300)
ax = axes[0]
tmp = gene_records.loc[gene_records['corr']>threspos]
sns.histplot(data=tmp, x='coord', y='corr', bins=300, ax=ax, cbar=True, cbar_kws=dict(ticks=[0,7], fraction=0.2, shrink=0.5))
ax.set_yticks([0.7, 0.8, 0.9, 1.0])
ax = axes[1]
# tmp = gene_records.loc[gene_records['corr']<thresneg]
tmp = gene_records.loc[gene_records['corr']<-threspos]
sns.histplot(data=tmp, x='coord', y='corr', bins=300, ax=ax, cbar=True, cbar_kws=dict(ticks=[0,3], fraction=0.2, shrink=0.5))
# ax.set_yticks([-0.6, -0.7, -0.8, -0.9])
ax.set_yticks([-0.7, -0.8, -0.9, -1.0])
ax.set_xticks([-300, -100, 100, 300])
ax.set_xticklabels(['-5M', 'TSS', 'TES', '+5M'])
# plt.savefig('DEG_domain_sigcorr.pdf', transparent=True)
[Text(-300, 0, '-5M'),
Text(-100, 0, 'TSS'),
Text(100, 0, 'TES'),
Text(300, 0, '+5M')]