DEG identification#
import numpy as np
import pandas as pd
import anndata
import scanpy as sc
indir = '/home/jzhou_salk_edu/sky_workdir/hba/rna_majortype/'
outdir = indir
gene_meta = pd.read_csv('/home/jzhou_salk_edu/sky_workdir/hba/ref/gencode.v33.basic.annotation.gene.flat.tsv.gz', sep='\t', index_col=8)
gene_meta
| chrom | source | feature | start | end | score | strand | phase | transcript_id | gene_type | ... | gene_name | transcript_type | transcript_status | transcript_name | exon_number | exon_id | level | hgnc_id | havana_gene | tag | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| gene_id | |||||||||||||||||||||
| ENSG00000223972.5 | chr1 | HAVANA | gene | 11869 | 14409 | . | + | . | NaN | transcribed_unprocessed_pseudogene | ... | DDX11L1 | NaN | NaN | NaN | NaN | NaN | 2 | HGNC:37102 | OTTHUMG00000000961.2 | NaN |
| ENSG00000227232.5 | chr1 | HAVANA | gene | 14404 | 29570 | . | - | . | NaN | unprocessed_pseudogene | ... | WASH7P | NaN | NaN | NaN | NaN | NaN | 2 | HGNC:38034 | OTTHUMG00000000958.1 | NaN |
| ENSG00000278267.1 | chr1 | ENSEMBL | gene | 17369 | 17436 | . | - | . | NaN | miRNA | ... | MIR6859-1 | NaN | NaN | NaN | NaN | NaN | 3 | HGNC:50039 | NaN | NaN |
| ENSG00000243485.5 | chr1 | HAVANA | gene | 29554 | 31109 | . | + | . | NaN | lncRNA | ... | MIR1302-2HG | NaN | NaN | NaN | NaN | NaN | 2 | HGNC:52482 | OTTHUMG00000000959.2 | ncRNA_host |
| ENSG00000284332.1 | chr1 | ENSEMBL | gene | 30366 | 30503 | . | + | . | NaN | miRNA | ... | MIR1302-2 | NaN | NaN | NaN | NaN | NaN | 3 | HGNC:35294 | NaN | NaN |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| ENSG00000198695.2 | chrM | ENSEMBL | gene | 14149 | 14673 | . | - | . | NaN | protein_coding | ... | MT-ND6 | NaN | NaN | NaN | NaN | NaN | 3 | HGNC:7462 | NaN | NaN |
| ENSG00000210194.1 | chrM | ENSEMBL | gene | 14674 | 14742 | . | - | . | NaN | Mt_tRNA | ... | MT-TE | NaN | NaN | NaN | NaN | NaN | 3 | HGNC:7479 | NaN | NaN |
| ENSG00000198727.2 | chrM | ENSEMBL | gene | 14747 | 15887 | . | + | . | NaN | protein_coding | ... | MT-CYB | NaN | NaN | NaN | NaN | NaN | 3 | HGNC:7427 | NaN | NaN |
| ENSG00000210195.2 | chrM | ENSEMBL | gene | 15888 | 15953 | . | + | . | NaN | Mt_tRNA | ... | MT-TT | NaN | NaN | NaN | NaN | NaN | 3 | HGNC:7499 | NaN | NaN |
| ENSG00000210196.2 | chrM | ENSEMBL | gene | 15956 | 16023 | . | - | . | NaN | Mt_tRNA | ... | MT-TP | NaN | NaN | NaN | NaN | NaN | 3 | HGNC:7494 | NaN | NaN |
60662 rows × 21 columns
adata = anndata.read_h5ad(f'{indir}rna_raw.h5ad')
adata
AnnData object with n_obs × n_vars = 29000 × 33387
obs: 'Class', 'Supercluster', 'Clusters', 'Age', 'Donor', 'Sex', 'Tissue', 'MajorType', 'Prob'
adata.var['ncell'] = adata.X.getnnz(axis=0)
adata = adata[:, (adata.var['ncell']>10) & (adata.var.index.isin(gene_meta.index))].copy()
adata.obs['TotalUMI'] = adata.X.sum(axis=1).A1
adata.X.data = adata.X.data / np.repeat(adata.obs['TotalUMI'].values, adata.X.getnnz(axis=1)) * adata.obs['TotalUMI'].median()
sc.pp.log1p(adata)
adata.var["mean"] = adata.X.mean(axis=0).A1
adata.var["std"] = (adata.X.multiply(adata.X)).mean(axis=0).A1 - (
adata.var["mean"].values ** 2
)
print(adata.var["std"].min())
3.7919453461654484e-05
leg = {'exc': ['L23_IT', 'L4_IT', 'L5_IT', 'L6_IT', 'L6_IT_Car3', 'L56_NP', 'L6_CT', 'L6b', 'L5_ET', 'Amy'],
'inh': ['Lamp5', 'Lamp5_LHX6', 'Sncg', 'Vip', 'Pvalb', 'Pvalb_ChC', 'Sst', 'CHD7'],
'msn': ['MSN_D1', 'MSN_D2', 'Foxp2'],
'sub': ['SubCtx'],
'glia': ['ASC', 'ODC', 'OPC'],
'mgc': ['MGC'],
'smc': ['PC'],
'endo': ['EC'],
'fibro': ['VLMC'],
}
leg['neu'] = leg['exc'] + leg['inh'] + leg['msn'] + leg['sub']
leg['all'] = leg['neu'] + leg['glia'] + leg['mgc'] + leg['smc'] + leg['endo'] + leg['fibro']
expr = pd.DataFrame([adata.X[adata.obs['MajorType']==xx].mean(axis=0).A.ravel() for xx in leg['all']], index=leg['all'], columns=adata.var.index)
data = {xx:adata.X[adata.obs['MajorType']==xx].toarray() for xx in leg['all']}
import os
from scipy.stats import ranksums, kruskal
from concurrent.futures import ProcessPoolExecutor, as_completed
from statsmodels.sandbox.stats.multicomp import multipletests as FDR
for group in ['all', 'neu', 'exc', 'inh']:
genefilter = (adata.X[adata.obs['MajorType'].isin(leg[group])].getnnz(axis=0) > 10)
stats = []
for i in range(adata.shape[1]):
if genefilter[i]:
tmp = [data[ct][:,i] for ct in leg[group]]
xx, yy = kruskal(*tmp)
stats.append([xx, yy])
stats = np.array(stats)
stats[:, 1] = FDR(stats[:, 1], 0.01, "fdr_bh")[1]
stats = pd.DataFrame(stats, columns=['stats', 'fdr'], index=adata.var.index[genefilter])
stats.to_hdf(f'{outdir}{group}_deg_stats.hdf', key='data')
print(group)
all
neu
exc
inh
leg = leg['all']
legname = ['L2/3-IT', 'L4-IT', 'L5-IT', 'L6-IT', 'L6-IT-Car3', 'L5/6-NP', 'L6-CT', 'L6b', 'L5-ET', 'Amy-Exc',
'Lamp5', 'Lamp5-Lhx6', 'Sncg', 'Vip', 'Pvalb', 'Pvalb-ChC', 'Sst', 'Chd7',
'MSN-D1', 'MSN-D2', 'Foxp2', 'SubCtx-Cplx',
'ASC', 'ODC', 'OPC', 'MGC', 'PC', 'EC', 'VLMC'
]
def diff_ranksum(c1, c2):
global data
# print(c1, c2)
fc = (data[c1].mean(axis=0) + 0.01) / (data[c2].mean(axis=0) + 0.01)
pv = np.zeros(len(fc))
for i in range(len(pv)):
pv[i] = ranksums(data[c1][:, i], data[c2][:, i])[1]
fdr = FDR(pv, 0.01, "fdr_bh")[1]
return [c1, c2, fc, fdr]
os.makedirs(f'{outdir}DEG/', exist_ok=True)
cpu = 32
with ProcessPoolExecutor(cpu) as executor:
futures = []
for c1 in range(len(leg)-1):
for c2 in range(c1+1, len(leg)):
future = executor.submit(
diff_ranksum,
c1=leg[c1],
c2=leg[c2],
)
futures.append(future)
result = []
for future in as_completed(futures):
result.append(future.result())
# c1, c2 = result[-1][0], result[-1][1]
# print(f'{leg[c1]} vs {leg[c2]} finished')
for xx in result:
np.savez(f'{outdir}DEG/{xx[0]}-{xx[1]}.npz', fc=xx[2], fdr=xx[3])
import seaborn as sns
import matplotlib as mpl
import matplotlib.pyplot as plt
mpl.style.use('default')
mpl.rcParams['pdf.fonttype'] = 42
mpl.rcParams['ps.fonttype'] = 42
mpl.rcParams['font.family'] = 'sans-serif'
mpl.rcParams['font.sans-serif'] = 'Helvetica'
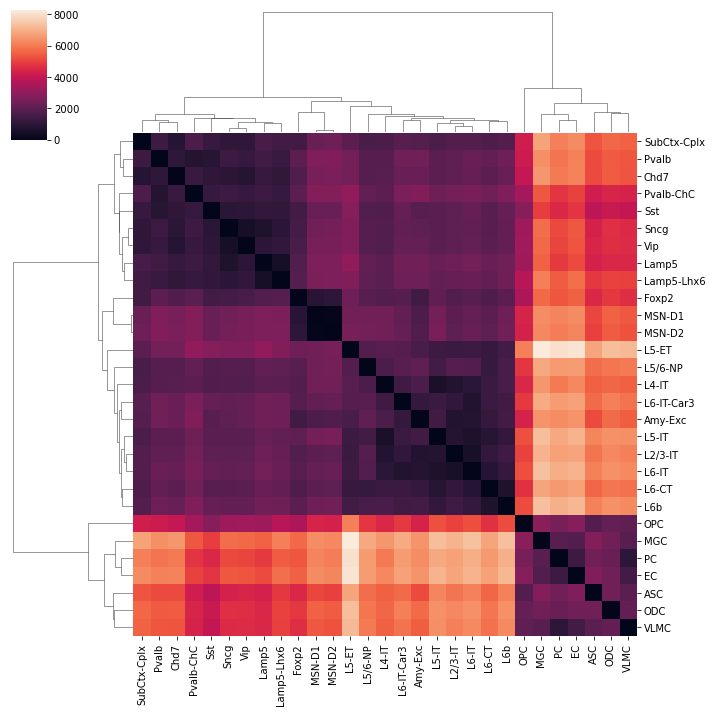
count = pd.DataFrame(index=leg, columns=leg).fillna(0)
for xx in result:
count.loc[xx[0], xx[1]] = np.sum(np.logical_and(np.abs(np.log2(xx[2]))>1, xx[3]<1e-3))
count = count + count.T
sns.clustermap(count, xticklabels=legname, yticklabels=legname)
<seaborn.matrix.ClusterGrid at 0x7f8e84712f50>
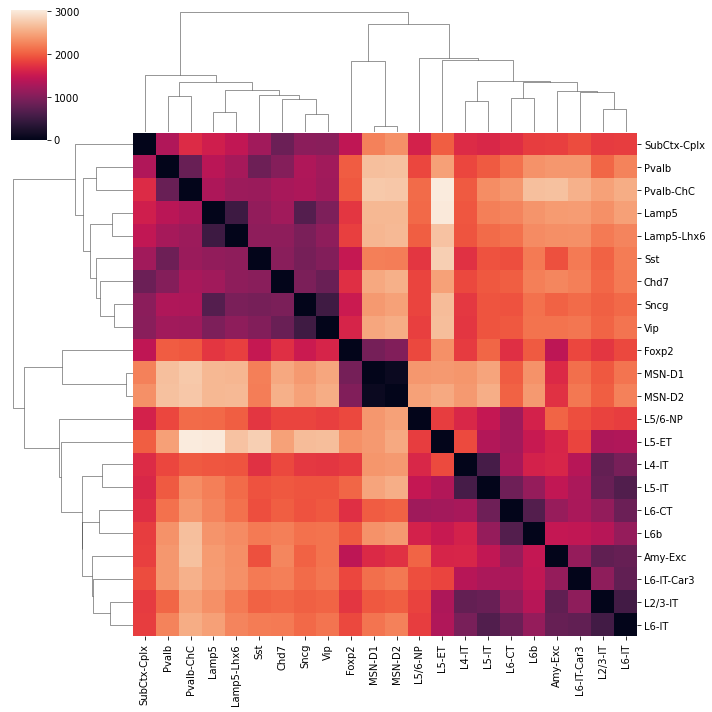
sns.clustermap(count.loc[leg[:22], leg[:22]], xticklabels=legname[:22], yticklabels=legname[:22])
<seaborn.matrix.ClusterGrid at 0x7f8e843de250>
from sklearn.preprocessing import normalize
from sklearn.decomposition import TruncatedSVD
from ALLCools.plot import *
from ALLCools.clustering import significant_pc_test, tsne
def dump_embedding(adata, name, n_dim=2):
# put manifold coordinates into adata.obs
for i in range(n_dim):
adata.obs[f"{name}_{i}"] = adata.obsm[f"X_{name}"][:, i]
return adata
model = TruncatedSVD(n_components=50)
data_reduce = model.fit_transform(adata.X)
data_reduce /= model.singular_values_
adata.obsm['pca_all'] = data_reduce.copy()
significant_pc_test(adata, p_cutoff=0.1, update=False, obsm="pca_all")
49 components passed P cutoff of 0.1.
49
npc = 50
adata.obsm['X_pca'] = normalize(data_reduce[:, :npc], axis=1)
tsne(
adata,
obsm="X_pca",
metric="euclidean",
exaggeration=-1,
perplexity=50,
n_jobs=-1,
)
dump_embedding(adata, "tsne")
adata.obsm[f"u{npc}_tsne"] = adata.obsm["X_tsne"].copy()
ds = 0.5
coord_base = 'tsne'
import hba_data
ctdict = hba_data.internal.celltype.CellType.majortype_palette()
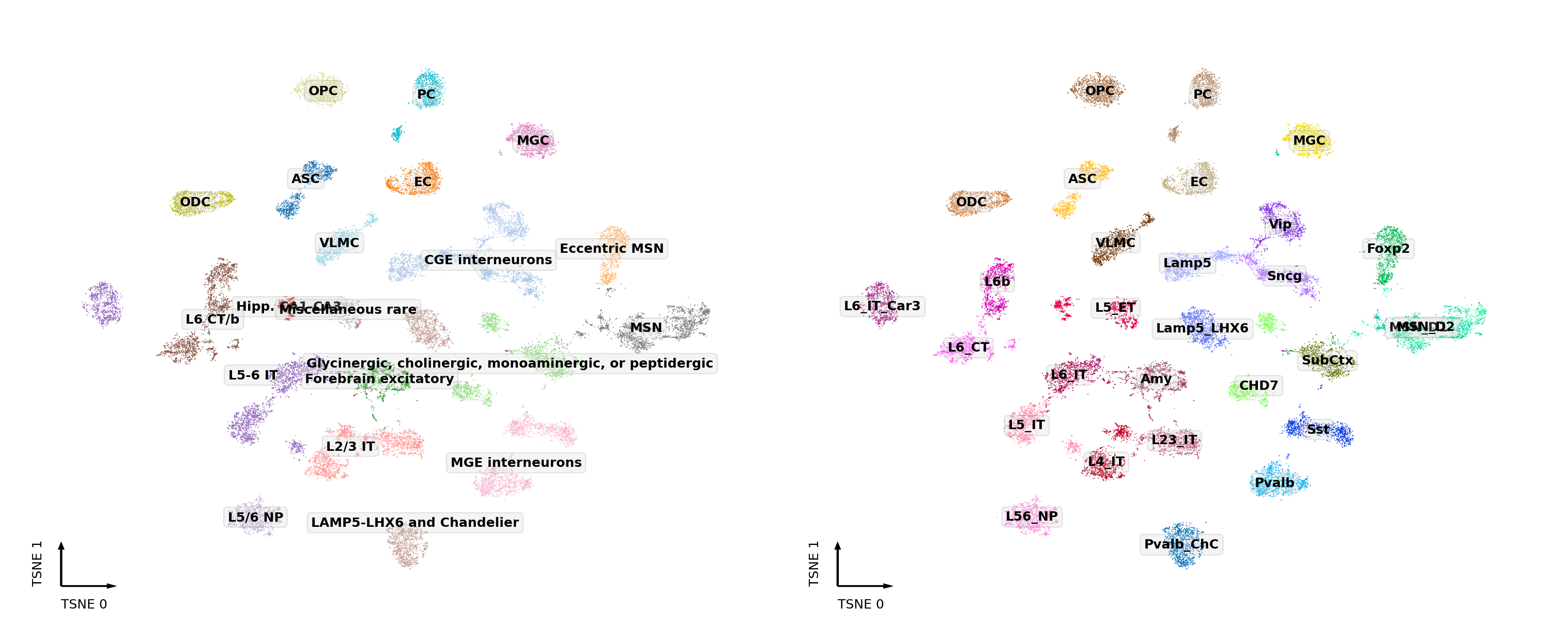
fig, axes = plt.subplots(1, 2, figsize=(10, 4), dpi=300, constrained_layout=True, sharex='all', sharey='all')
ax = axes[0]
tmp = adata.obs['Supercluster'].value_counts()
tmp = adata.obs.loc[adata.obs['Supercluster'].isin(tmp.index[tmp>50])]
_ = categorical_scatter(data=tmp,
ax=ax,
coord_base=coord_base,
s=ds,
hue='Supercluster',
text_anno='Supercluster',
palette='tab20',
labelsize=6,
max_points=None,
scatter_kws={'rasterized':True},
#show_legend=True,
#legend_kws={'ncol':1},
)
ax = axes[1]
_ = categorical_scatter(data=adata.obs,
ax=ax,
coord_base=coord_base,
s=ds,
hue='MajorType',
text_anno='MajorType',
palette=ctdict,
labelsize=6,
max_points=None,
scatter_kws={'rasterized':True},
#show_legend=True,
#legend_kws={'ncol':1},
)
findfont: Font family ['sans-serif'] not found. Falling back to DejaVu Sans.
findfont: Generic family 'sans-serif' not found because none of the following families were found: Helvetica
findfont: Font family ['sans-serif'] not found. Falling back to DejaVu Sans.
findfont: Generic family 'sans-serif' not found because none of the following families were found: Helvetica
adata.var[['gene_name', 'chrom', 'start', 'end']] = gene_meta.loc[adata.var.index, ['gene_name', 'chrom', 'start', 'end']].values
adata.var[['start', 'end']] = adata.var[['start', 'end']].astype(int)
adata.write_h5ad(f'{outdir}cell_{adata.shape[0]}_rna.h5ad')
from scipy.stats import rankdata
selg = np.zeros(adata.shape[1])
for xx in result:
if (xx[0] in leg[:22]) and (xx[1] in leg[:22]):
rank = rankdata(xx[3])
selg[rank<=100] = 1
print(selg.sum())
1173.0
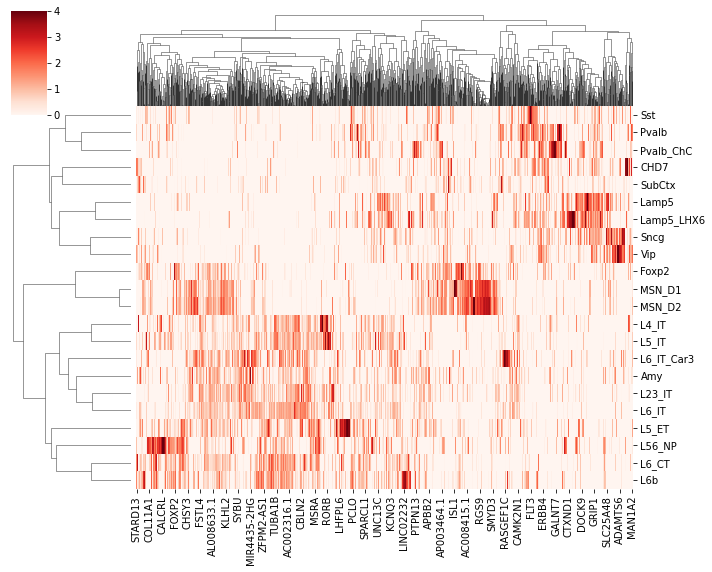
tmp = expr.loc[leg[:22], selg==1]
tmp.columns = adata.var['gene_name'].values[selg==1]
sns.clustermap(tmp, z_score=1, cmap='Reds', vmin=0, vmax=4, metric='cosine', figsize=(10,8))
<seaborn.matrix.ClusterGrid at 0x7f8dc6837510>
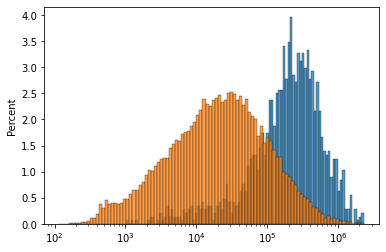
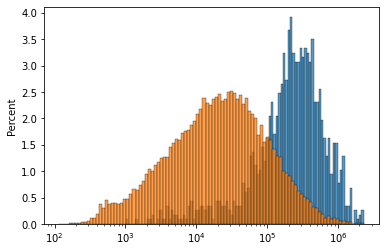
fig, ax = plt.subplots()
sns.histplot(adata.var.loc[selg==1, 'end'] - adata.var.loc[selg==1, 'start'], log_scale=10, bins=100, stat='percent', ax=ax)
sns.histplot(adata.var['end'] - adata.var['start'], log_scale=10, bins=100, stat='percent', ax=ax)
<AxesSubplot:ylabel='Percent'>
expr.to_hdf(f'{outdir}cluster_expr.hdf', key='data')
selg = np.zeros(adata.shape[1])
for xx in result:
rank = rankdata(xx[3])
selg[rank<=100] = 1
print(selg.sum())
1439.0
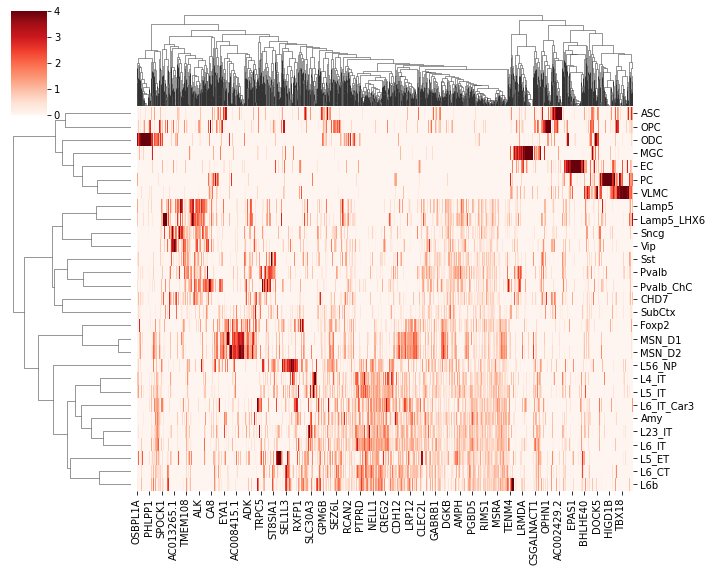
tmp = expr.loc[leg, selg==1]
tmp.columns = adata.var['gene_name'].values[selg==1]
sns.clustermap(tmp, z_score=1, cmap='Reds', vmin=0, vmax=4, metric='cosine', figsize=(10,8))
<seaborn.matrix.ClusterGrid at 0x7f8dc6503f10>
fig, ax = plt.subplots()
sns.histplot(adata.var.loc[selg==1, 'end'] - adata.var.loc[selg==1, 'start'], log_scale=10, bins=100, stat='percent', ax=ax)
sns.histplot(adata.var['end'] - adata.var['start'], log_scale=10, bins=100, stat='percent', ax=ax)
<AxesSubplot:ylabel='Percent'>